About us
Overview

Cell division is one of the most fundamental processes in biology. Thousands of proteins have to be made successfully to enable accurate duplication of the genetic information, segregate duplicated chromosomes into daughter cells and to complete cell division. Our lab aims to provide important new insights into how the genome can be duplicated with high accuracy, how damage to the genome can be recognised and repaired and what consequences arise if these processes fail. Our research will provide advances of our fundamental understanding of cell biology. However, we aim to explore how new insights can be exploited towards diagnosing and treating human disease, with a particular focus on cancer. We use a range of advanced molecular cell biology and genetics techniques for traditional as well as new experimental approaches. Our research has been funded by The Leverhulme Trust and the Royal Society. Currently our work is funded by the BBSRC.
Research
DNA encodes the information that provides the basis for all life. For cell division to take place the entire DNA of a cell has to be fully duplicated, ensuring that both the mother and the daughter cell can get one complete copy. In addition, the copy made has to be identical to the original. Any changes to the DNA can potentially be harmful, as they will alter how cells function, or even lead to cell death. On a single cell level, accuracy of the duplication process is normally so high that not a single error is made when the millions of DNA base pairs are copied. This extraordinarily high level of accuracy is achieved by a network of different processes that regulate the duplication process and choreograph segregation of the two complete copies into mother and daughter cells. However, mutations in the DNA can inactivate this network of processes and it is a hallmark of cancer cells that they grow in an uncontrolled way.
Our lab is interested in how the duplication process can reach such high levels of accurracy and currently we are using the bacterium Escherichia coli as a model to study all stages of DNA replication, cell cycle control and genomic stability. Like many other bacteria, E. coli has a single chromosome. Replication of DNA is initiated at one specific site called the origin of replication (oriC).
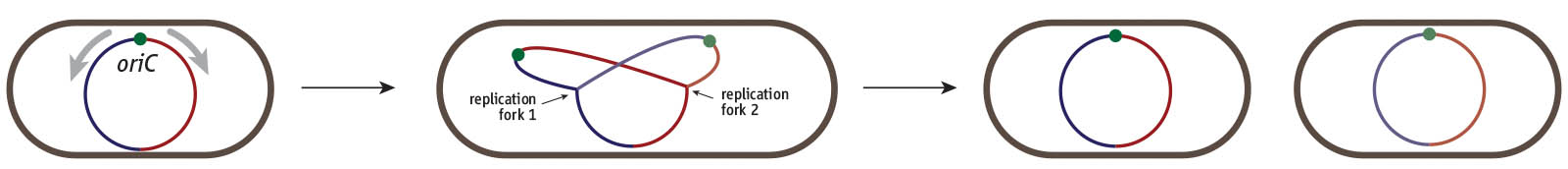
Cell division cycle of a bacterium such as E. coli. Cells harbour a single circular chromosome which contains a single replication origin called oriC. Two replication forks are recruited and duplicate the chromosome in opposite directions, thereby dividing the chromosome into a clockwise (red) and counterclockwise (blue) half or replichore. Once replication is complete the two chromosomes can be segregated into the two daughter cells.
Two complex machines termed replisomes are recruited to this origin and then move in opposite directions, each copying a chromosomal half with high speed and precision. Replication continues until one fork meets another coming the opposite way.
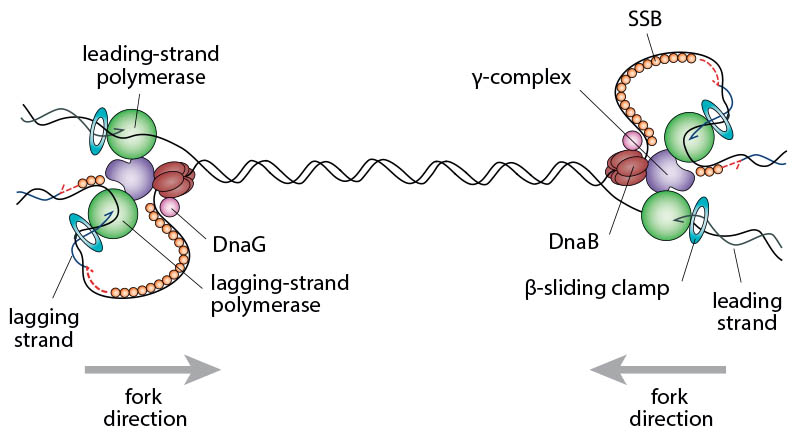
Schematic illustrating the complexity of two replication forks converging opposite the origin. In order to complete chromosomal replication the DNA between the replisomes has to be fully replicated.
Currently we are focussing on the final stage of DNA replication, which is called replication termination. Our research has revealed that the collision of two fast moving replication forks has the potential to corrupt the DNA by provoking recombination events. This cause of mutation is unexpected, because the fusion of two such replication forks is a fundamental process when DNA is copied, and we have identified a number of pathways that can prevent the harmful consequences of fork fusions. In E. coli the fact that replication is carried out by two replication forks dictates that only one such fusion event occurs. In human cells the duplication process is initiated at hundreds of origins, leading to hundreds of fusion events, making studies much more complex. The relative simplicity of fork fusions in E. coli will allow us to study these events in great detail with many different tools. Our studies will allow us to generate a model of how the duplication process of DNA is normally brought to an orderly completion, which will help us to understand this process in our own cells, and our work will reveal whether it might contribute to the development of cancer and ageing.
In order to measure and visualise different aspects of DNA replication and DNA repair, we use a wide variety of tools and techniques.
Genetics
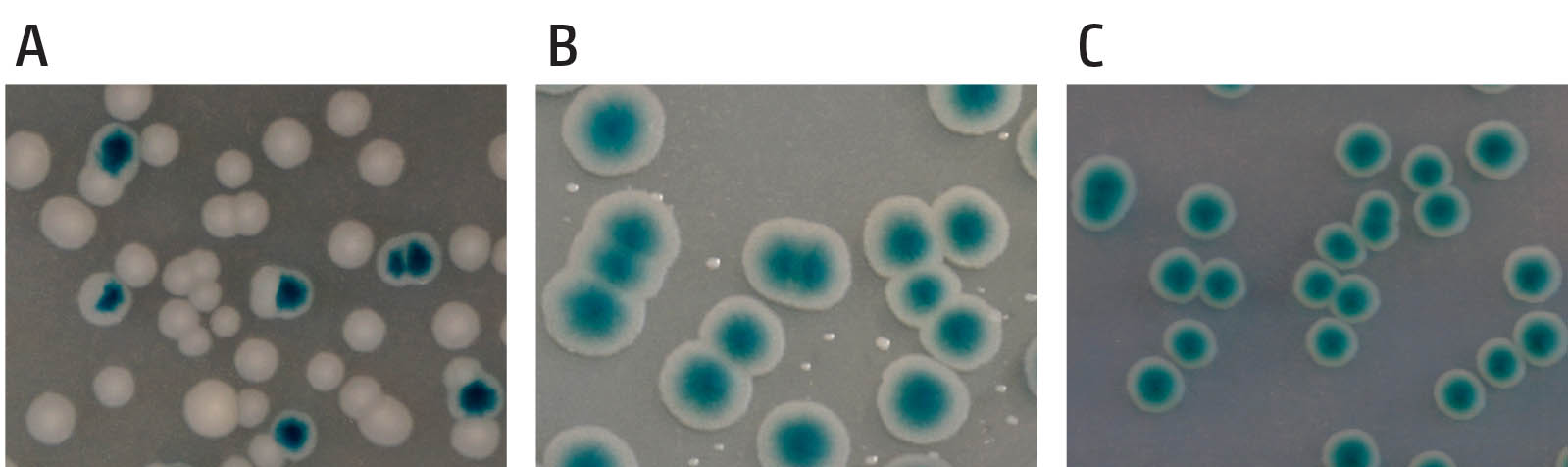
Example of the synthetic lethality screen. (A) shows cells in which the gene encoded on the pRC7 is not needed. Segregation of blue and white colonies is observed and blue colonies contain white sectors, demonstrating the rapid loss of the covering plasmid. (B) shows a synthetically sick interaction of two genes of interest. Cells contain a deletion of two genes, which renders the cells sick and very slow growing. Cells retaining the covering plasmid show rapid growth into unsectored blue colonies, as one of the two deletions is masked, while cells having lost the plasmid show very small white colonies, demonstrating that they are viable but very slow growing. (C) shows a synthetically lethal interaction of a deletion of two genes. Cells which have lost the covering plasmid are entirely unable to grow and no white colonies are observed, while cells retaining the covering plasmid grow into unsectored blue colonies.
We use traditional genetics approaches for strain constructions and phenotypic investigations, such as P1vir transduction, single-step gene disruption and many others. We routinely perform phenotypic analyses such as doubling time, sensitivity to damage induced by a variety of genotoxic agents, delay of cell division following DNA damage, growth in the absence of an active origin etc. In addition, a synthetic lethality assay is particularly informative to assess the genetic interactions of genes and point mutations. This assay employs the unstable pRC7 plasmid, which is rapidly lost, with cloned genes of interest to cover for chromosomal deletions. pRC7 carries a functional lac operon, and its loss can be revealed in a lac– mutant background on plates containing the beta-galactosidase indicator X-Gal by the formation of white colonies or white sectors (lac–) within blue colonies, depending on whether plasmid loss occurred before or after plating. This assay can efficiently highlight synthetically lethal but also synthetically sick genetic interactions.
Visualisation of protein and chromosome dynamics
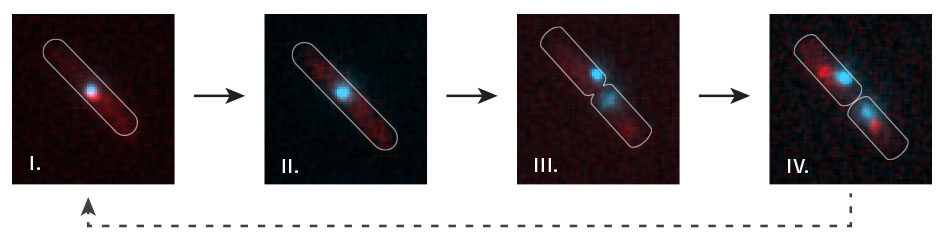
Time lapse visualisation of replication fork dynamics and terminus segregation in E. coli. The terminus region of the chromosome (cyan; visualised by lac repressor-fluorescent fusion proteins bound to lac operators) co-localises with replication forks (red focus) as it is replicated (i). All forks are disassembled at the termination area (ii – the red focus disappears) prior to chromosome segregation (iii – two cyan labelled termini appear). New replication forks are subsequently assembled (iv – a red focus in each of the daughter cells).
For cell biological visualisations we use a number of modern tools. Protein dynamics at replication forks in living cells can be visualised by fusing proteins of interest to fluorescent proteins. This allows us to study localisations within the cell, but also protein dynamics as complexes might be assembled and disassembled. In addition, chromosome dynamics can be visualised using fluorescent repressor-operator systems (FROS), allowing the visulisation of the location of defined chromosomal areas. If combined together, both systems provide a measure of when active replisomes reach defined areas of the chromosome, enabling us to specifically study the events that occur when replisomes reach the termination area of the chromosome.
Molecular Cell Biology
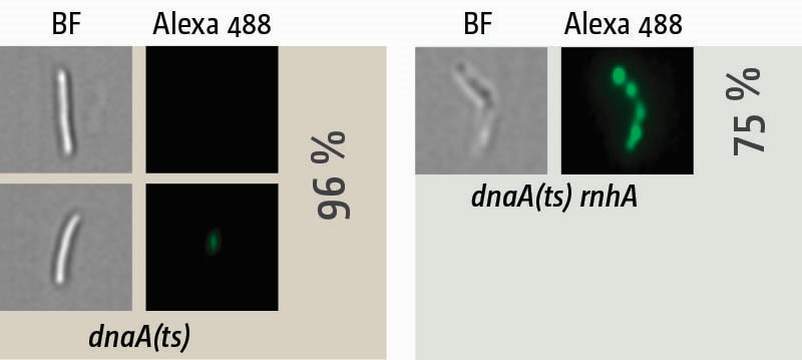
Visualisation of synthesis in cells in which oriC-firing is inhibited via a temperature-sensitive allele of the DnaA initiator protein (dnaA(ts)). Cells were shifted to restrictive temperature for 90 min, allowing all ongoing replication forks to complete synthesis while new forks at oriC cannot be established. Cells were then pulse-labelled with EdU for 15 min and, following the click-labelling reaction, visualised with an ImageStreamX Mark II. In dnaA(ts) single mutants no or very little synthesis is observed, while cells lacking RNase HI show robust level of synthesis in 75% of cells, providing an explanation why cells lacking RNase HI are capable of surviving without an active replication origin.
The molecular biology of DNA replication can not only be measured but also visualised by the incorporation of nucleotide analogues into nascent DNA. Incorporation of BrdU with subsequent separation of NotI-cut fragments of the chromosome via pulsed-field gel electrophoresis allows the visualisation of chromosomal areas with ongoing DNA synthesis in a population of cells. However, we can also visualise ongoing synthesis on a single cell level. By pulse-labeling newly replicated DNA with 5-ethynyl-2-deoxyuridine (EdU) followed by a click chemistry reaction and high-resolution microscopy in flow with an Amnis ImageStreamX Mark II, which combines the power of FACS analysis with single cell visualisation, we can quantify the amount of label while simultaneously imaging the location of where labelling is taking place (Dimude et al., 2015).
Replication profiles at a high resolution
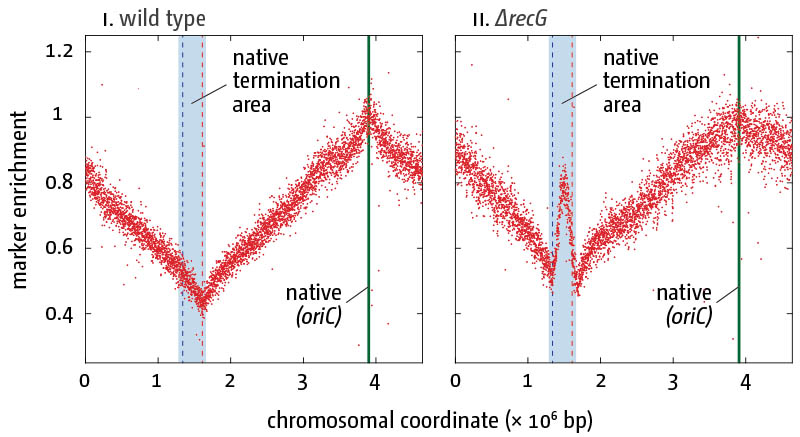
Replication profiles in E. coli cells in the presence (i) and absence (ii) of RecG helicase/tranlocase. Profiling of exponential-phase cells normalized against a stationary wild type culture are shown. The genome-wide high-resolution marker frequency analysis reveals high level of over-replication in the termination area of the chromosome in cells lacking RecG.
To visualise copy number variations of defined chromosomal areas, a variety of tools are used. qPCR is suitable to analyse relative abundance of defined chromosomal areas. To investigate whole chromosome replication dynamics, high-resolution replication profiles generated via Deep Sequencing can be used. This marker frequency analysis measures the duplication of each part of the genome as an increase in copy number following replication. It can very efficiently reveal aberrant processing of replication intermediates via under- and over-representation of specific DNA sequences. For example, it has allowed us to demonstrate that cells lacking RecG helicase/translocase show significant levels of over-replication specifically in the termination area of the chromosome. Our experiments have identified RecG, together with 3' exonucleases, as key proteins required for the processing of fork fusion intermediates to prevent termination-induced over-replication.
Rudolph BLOG
pE-4000 by CoolLED
Switching from metal halide to LED was a very good move for us.
Older articles
People
Lab members




Undergraduate and MSc students

Brian Prince-Lombart
Previous lab members

Dr Sarah L. Midgley-Smith

Dr. Amy Upton

Darja Ivanova

Toni Taylor

Monja Stein

Mohammad Khalifa

Sabiha Karim

Vladyslava Borysenko

Ewa Andrzejewska

Dominika Warecka

Anastasia Georgievskaya

Alexandra Gajdosova
Publications
Great stuff we wrote.
- Goodall D.J., Warecka D., Hawkins M. and C.J. Rudolph (2023). Interplay between chromosomal architecture and termination of DNA replication in bacteria. Front. Microbiol 14. DOI: 10.3389/fmicb.2023.1180848. [Link]
- Killelea T., Dimude J.U., He L., Stewart A.L., Kemm F.E., Radovčić M., Ivančić-Baće I., Rudolph C.J. and E.L. Bolt (2023). Cas1-Cas2 physically and functionally interacts with DnaK to modulate CRISPR adaptation. Nucleic Acids Research, gkad473, DOI: 10.1093/nar/gkad473 [Link]
- de Dios R., Proctor C.R., Maslova E., Dzalbe S., Rudolph C.J. and Ronan R. McCarthy (2023). Artificial sweeteners inhibit multidrug resistant pathogen growth and potentiate antibiotic activity. EMBO Mol. Med. 15(1):e16397. DOI: 10.15252/emmm.202216397. [Link]
- Jameson K.H., Rudolph C.J. and M. Hawkins (2021). Termination of DNA replication at Tus-ter barriers results in under-replication of template DNA. J Biol Chem. 297 (6), 101409. doi: 10.1016/j.jbc.2021.101409 [Link]
- Goodall D.J., Jameson K.H., Hawkins M. and C.J. Rudolph (2021). A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups. Int. J. Mol. Sci. 22 (15), 7928. DOI: 10.3390/ijms22157928. [Link]
- Syeda A.H., Dimude J.U., Skovgaard O. and C.J. Rudolph (2020). Too Much of a Good Thing: How Ectopic DNA Replication Affects Bacterial Replication Dynamics. Front. Microbiol., 15 April 2020. doi: 10.3389/fmicb.2020.00534 [Link]
- Hawkins M., Dimude J.U., Howard J.A.L., Smith A.J., Dillingham M.S., Savery N.J., Rudolph C.J. and P. McGlynn (2019). Direct removal of RNA polymerase barriers to replication by accessory replicative helicases. Nucleic Acids Res 47(10):5100–5113. [Link]
- Midgley-Smith, S.L.; Dimude, J.U.; Taylor, T.; Forrester, N.M.; Upton, A.L.; Lloyed, R.G. and C. J. Rudolph (2018). Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res. 46(15):7701-7715. [Link]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A. and R.G. Lloyd (2013). Avoiding chromosome pathology when replication forks collide. Nature 500 (7464), 608–11. [Link]
- Midgley-Smith, S.L., Dimude, J.U. and C.J. Rudolph (2018). A role for 3' exonucleases at the final stages of chromosome duplication in Escherichia coli. Nucleic Acids Res. 47(4):1847–1860 [Link]
- Rudolph C.J., Corocher T.-A., Grainge I. and I.G. Duggin (2019). Termination of DNA Replication in Prokaryotes. Wiley electronic Library of Science (eLS). DOI: 10.1002/9780470015902.a0001056.pub3 [Link]
- Dimude, J.U., Midgley-Smith, S.L. and C.J. Rudolph (2018). Replication-transcription conflicts trigger extensive DNA degradation in Escherichia coli cells lacking RecBCD. DNA Repair (Amst) 70:37–48. [Link]
- Dimude, J.U.; Stein, M.; Andrzejewska, E.E.; Khalifa, M.S.; Gajdosova, A.; Retkute, R.; Skovgaard, O. and C.J. Rudolph (2018). Origins Left, Right, and Centre: Increasing the Number of Initiation Sites in the Escherichia coli Chromosome. Genes 9 (8), pii: E376. [Link]
- Dimude, J.U.; Midgley-Smith, S.L.; Stein, M. and Rudolph, C.J. (2016). Replication Termination: Containing Fork Fusion-Mediated Pathologies in Escherichia coli. Genes 7(8), 40; doi:10.3390/genes7080040. [Link]
- Lloyd, R.G. and C.J. Rudolph (2016). 25 years on and no end in sight: a perspective on the role of RecG protein. Curr. Genet. 62(4):827–40. doi: 10.1007/s00294-016-0589-z [Link]
- Dimude, J.U.; Stockum, A; Midgley-Smith, S.L.; Upton, A.L.; Foster, H.A.; Khan, A; Saunders, N.J.; Retkute, R. and C.J. Rudolph (2015). The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. mBio. 6(6). pii: e01294-15. doi: 10.1128/mBio.01294–15. [Link]
- Ivanova, D.; Taylor, T.; Smith, S.L.; Dimude, J.U.; Upton, A.L.; Mehrjouy, M.M.; Skovgaard, O.; Sherratt, D.J.; Retkute, R. and C.J. Rudolph (2015). Shaping the landscape of the Escherichia coli chromosome: replication-transcription encounters in cells with an ectopic replication origin. Nucleic Acids Res. 43(16):7865–77. [Link]
- Upton, A.L.; Grove, J.I.; Mahdi, A.A.; Briggs, G.S.; Milner, D.S.; Rudolph, C.J. and R.G. Lloyd (2014). Cellular location and activity of Escherichia coli RecG proteins shed light on the function of its structurally unresolved C-terminus. Nucleic Acids Res. 42 (9), 5702–14. [Link]
- Stockum, A.; Lloyd, R.G. and C.J. Rudolph (2012). On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiology 12:26. [Link]
- Atkinson, J.; Gupta, M.K.; Rudolph, C.J.; Bell, H.; Lloyd, R.G. and P. McGlynn (2011). Localisation of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res 39 (3), 949–957. [Link]
- Ede C, Rudolph CJ, Lehmann S, Schürer KA, Kramer W. (2011). Budding yeast Mph1 promotes sister chromatid interactions by a mechanism involving strand invasion. DNA Repair 10 (1), 45–55 [Link]
- Rudolph C.J., Mahdi A.A., Upton A.L. and R.G. Lloyd (2010). Single-strand DNA exonucleases protect Escherichia coli cells from the pathological consequences of unscheduled DNA replication. Genetics 186 (2), 473–492. [Link]
- Rudolph C.J., Upton A.L., Briggs, G.S. and R.G. Lloyd (2010). Is RecG a general guardian of the bacterial genome? DNA Repair 9 (3), 210–223. [Link]
- Guy C.P., Atkinson J., Gupta M.K., Mahdi A.A., Gwynn E.J., Rudolph C.J., Moon P.B., van Knippenberg I.C., Cadman C.J., Dillingham M.S., Lloyd R.G. and McGlynn P. (2009). Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36 (4), 654–666. [Link]
- Rudolph C.J., Upton A.L. and R.G. Lloyd (2009). Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 74 (4), 940–955. [Link]
- Rudolph C.J., Upton A.L., Harris L. and R.G. Lloyd (2009). Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73 (3), 352–366. [Link]
- Rudolph C.J., Upton A.L. and R.G. Lloyd (2008). Maintaining replication fork integrity in UV-irradiated Escherichia coli cells. DNA Repair 7 (9), 1589–1602. [Link]
- Rudolph C.J., Dhillon P., Moore T. and R.G. Lloyd (2007). Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair 6 (7), 981–993. [Link]
- Rudolph C.J., Upton A.L. and R.G. Lloyd (2007). Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev 21, 668–681. [Link]
Find us
We are always interested in motivated students with biology, genetics or biochemstry backgrounds and enquiries with CV are welcome.
Rudolph LAB
![]() Division of Bioscience
Division of Bioscience
Heinz Wolff Building Room 247
Brunel University London
Kingston Lane
Uxbridge, UB8 3PH
UK